I am a Principal Researcher at Microsoft Research New England. I also maintain a faculty position in the School of Public Health as an Associate Professor of Biostatistics with an affiliation in the Center for Computational Molecular Biology at Brown University. The central aim of my research program is to build machine learning algorithms and statistical tools that aid in the understanding of how nonlinear interactions between genetic features affect the architecture of complex traits and contribute to disease etiology. An overarching theme of the research done in the Crawford Lab group is to take modern computational approaches and develop theory that enable their interpretations to be related back to classical genomic principles. Some of my most recent work has landed me a place on Forbes 30 Under 30 list and recognition as a member of The Root 100 Most Influential African Americans in 2019. I have also been fortunate enough to be awarded an Alfred P. Sloan Research Fellowship, a David & Lucile Packard Foundation Fellowship for Science and Engineering, and a COPSS Emerging Leader Award.
Prior to joining both MSR and Brown, I received my PhD from the Department of Statistical Science at Duke University where I was co-advised by Sayan Mukherjee and Kris C. Wood. As a Duke Dean’s Graduate Fellow and NSF Graduate Research Fellow I completed my PhD dissertation entitled "Bayesian Kernel Models for Statistical Genetics and Cancer Genomics" which was awarded a Leonard J. Savage Award in Applied Methodology. I also received my Bachelors of Science degree in Mathematics from Clark Atlanta University.
Interpretability in Machine Learning Methods
Machine learning algorithms have become frequently used in genomic studies because they typically exhibit high predictive accuracy. However, recently, these same algorithms have also become criticized as “black box” techniques. We look to build methods that over this challenge.
Dissecting Genetic Architecture of Complex Traits
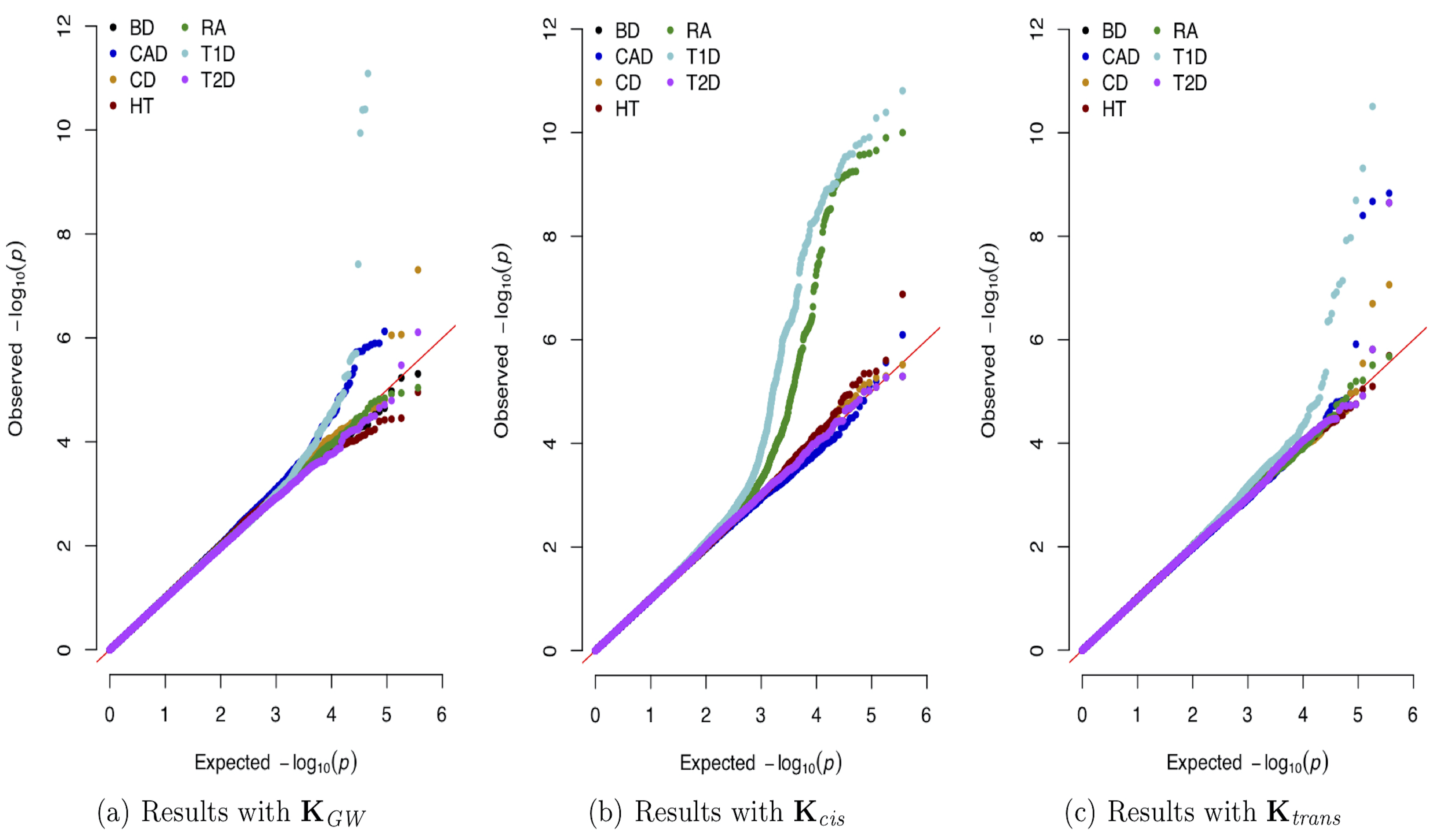
The explosion of large-scale genomic datasets has provided the unique opportunity to move beyond the traditional LMM framework within GWAS. We build novel ML methods that exhibit power for complex traits that are driven by non-additive genetic variation (e.g., gene-by-gene interactions).
Modeling 3D Variation with Topological Summaries
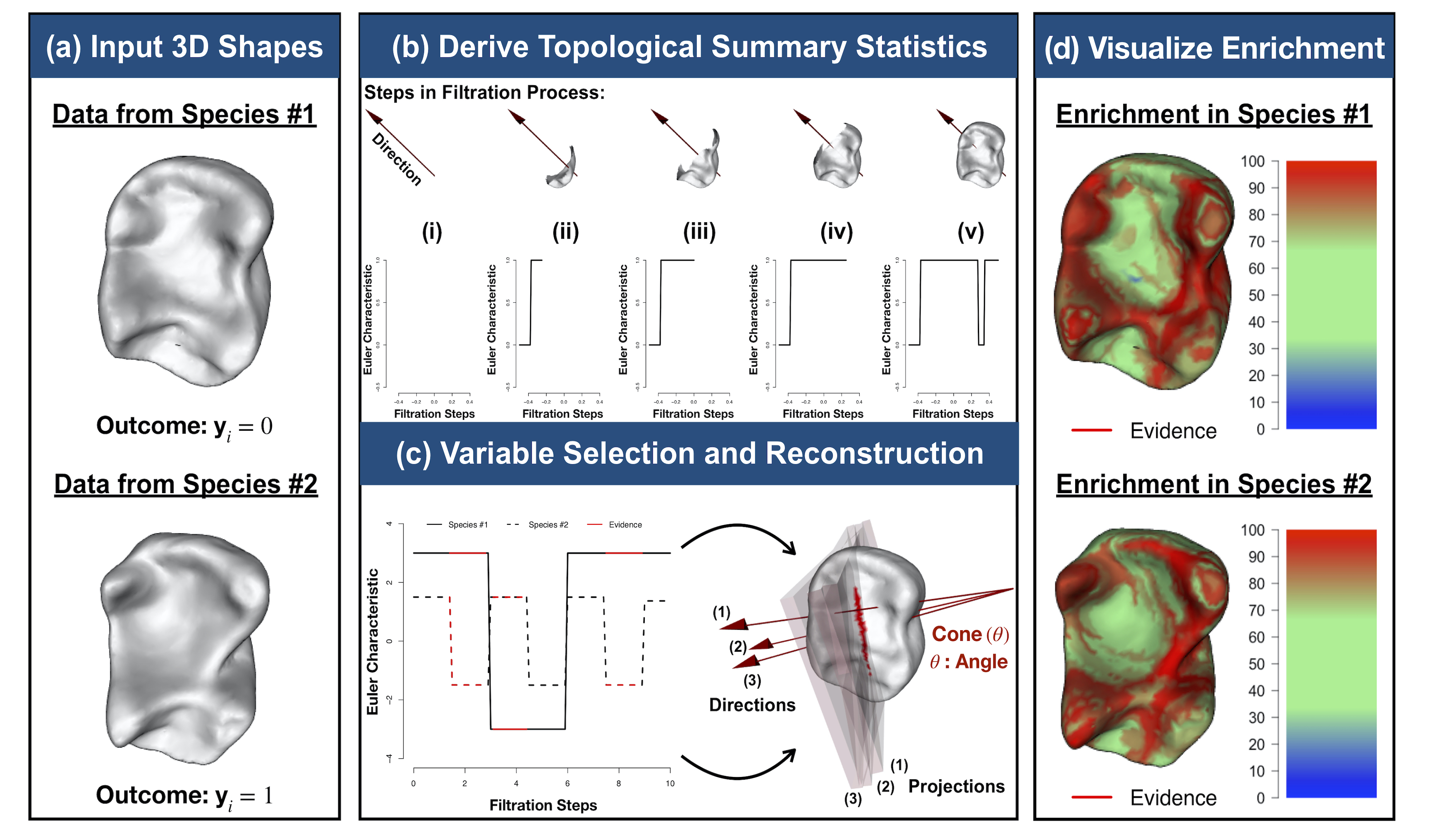
It has been a longstanding challenge to implement an analogue of variable selection with 3D shapes as the covariates in a regression model. Here, we develop novel statistical and topological data analytic (TDA) pipelines for sub-image selection where the goal is to identify the physical features of 3D shapes that best explain the variation between two phenotypic classes.
Statistical Methods for Cancer Pharmacology
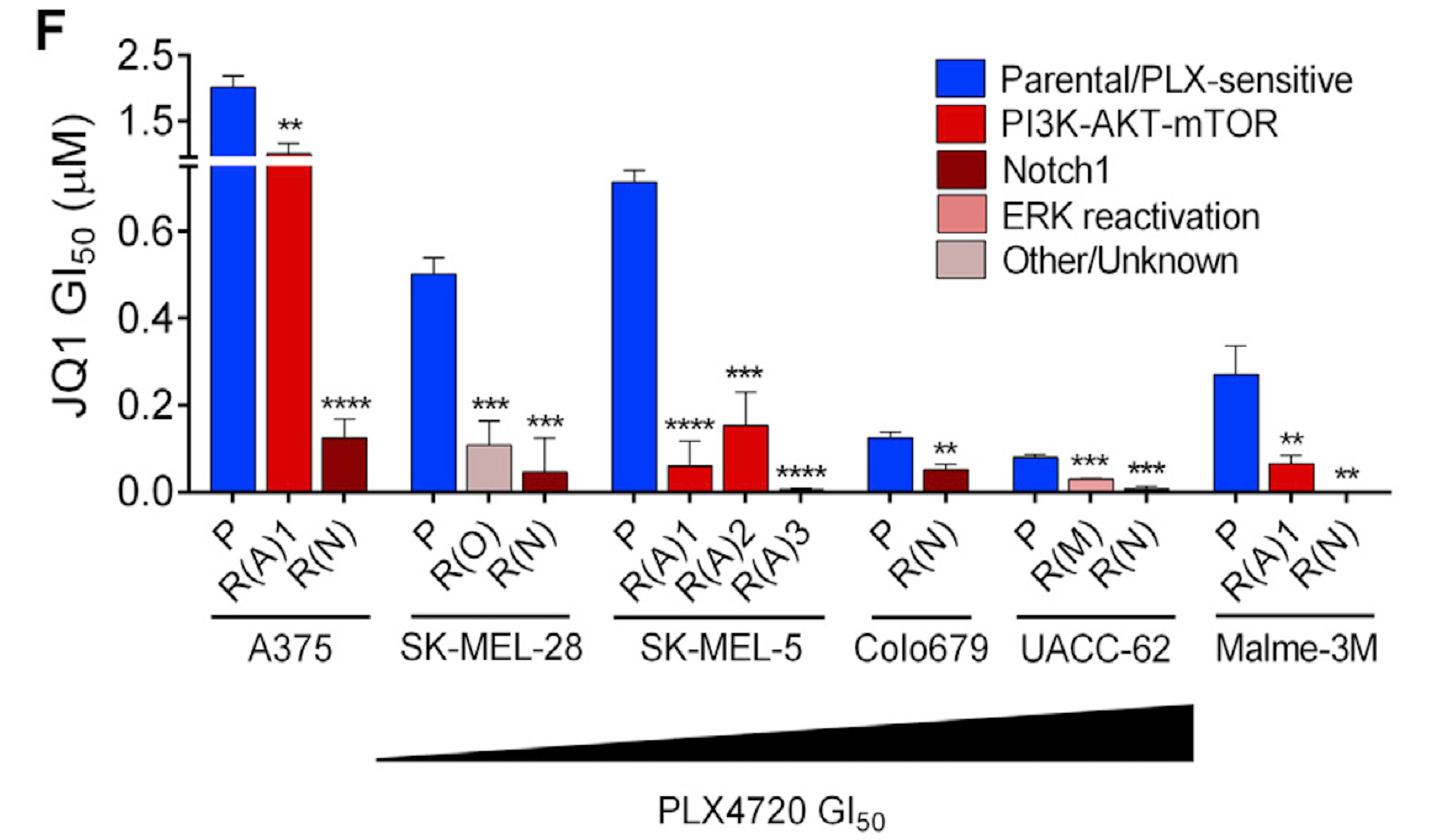
Targeted therapies aimed to inhibit oncogenic signaling within many cancer subtypes have been proven to have high initial clinical responses, but relapse in these patients is almost inevitable. To better understand this phenomenon, we develop algorithms that define rigorous transcriptional signatures of cancer recurrence and therapeutic resistance.
Publications Key: * co-first authors; † co-senior authors; # corresponding author(s); advisee
Preprint(s):
- A. DenAdel, M.L. Ramseier, A. Navia, A.K. Shalek, S. Raghavan, P.S. Winter, A.P. Amini, and L. Crawford#. A knockoff calibration method to avoid over-clustering in single-cell RNA-sequencing. bioRxiv. 2024.03.08.584180. [Preprint] [Software] [Documentation]
- C. Nwizu, M. Hughes, M.L. Ramseier, A. Navia, A.K. Shalek, N. Fusi, S. Raghavan†, P.S. Winter†, A.P. Amini†#, and L. Crawford†#. Scalable nonparametric clustering with unified marker gene selection for single-cell RNA-seq data. bioRxiv. 2024.02.11.579839. [Preprint] [Software] [Documentation]
- E.T. Winn-Nuñez#, H. Witt, D. Bhaskar, R.Y. Huang, J.S. Reichner, I.Y. Wong, and L. Crawford#. Generative modeling of biological shapes and images using a probabilistic α-shape sampler. bioRxiv. 2024.01.09.574919. [Preprint] [Software]
- K.Z. Kedzierska#, L. Crawford†, A.P. Amini†, and A.X. Lu†#. Assessing the limits of zero-shot foundation models in single-cell biology. bioRxiv. 2023.10.16.561085. [Preprint] [Software]
- H. Xie, L. Crawford#, and A. Conard#. Multioviz: an interactive platform for in silico perturbation and interrogation of gene regulatory networks. bioRxiv. 2023.10.10.561790. [Preprint] [Software] [Online Tool]
- K. Meng#, M. Ji, J. Wang, K. Ding, H. Kirveslahti, A. Eloyan, and L. Crawford. Statistical inference on grayscale images via the Euler-Radon transform. arXiv. 2308.14249. [Preprint] [Software]
- S.P. Smith*, G. Darnell*, D. Udwin, A. Harpak, S. Ramachandran†, and L. Crawford†#. Accounting for statistical non-additive interactions enables the recovery of missing heritability from GWAS summary statistics. bioRxiv. 2022.07.21.501001. [Preprint] [Software]
- K. Meng#, J. Wang, L. Crawford, and A. Eloyan. Randomness and statistical inference of shapes via the smooth Euler characteristic transform. arXiv. 2204.12699. [Preprint] [Software]
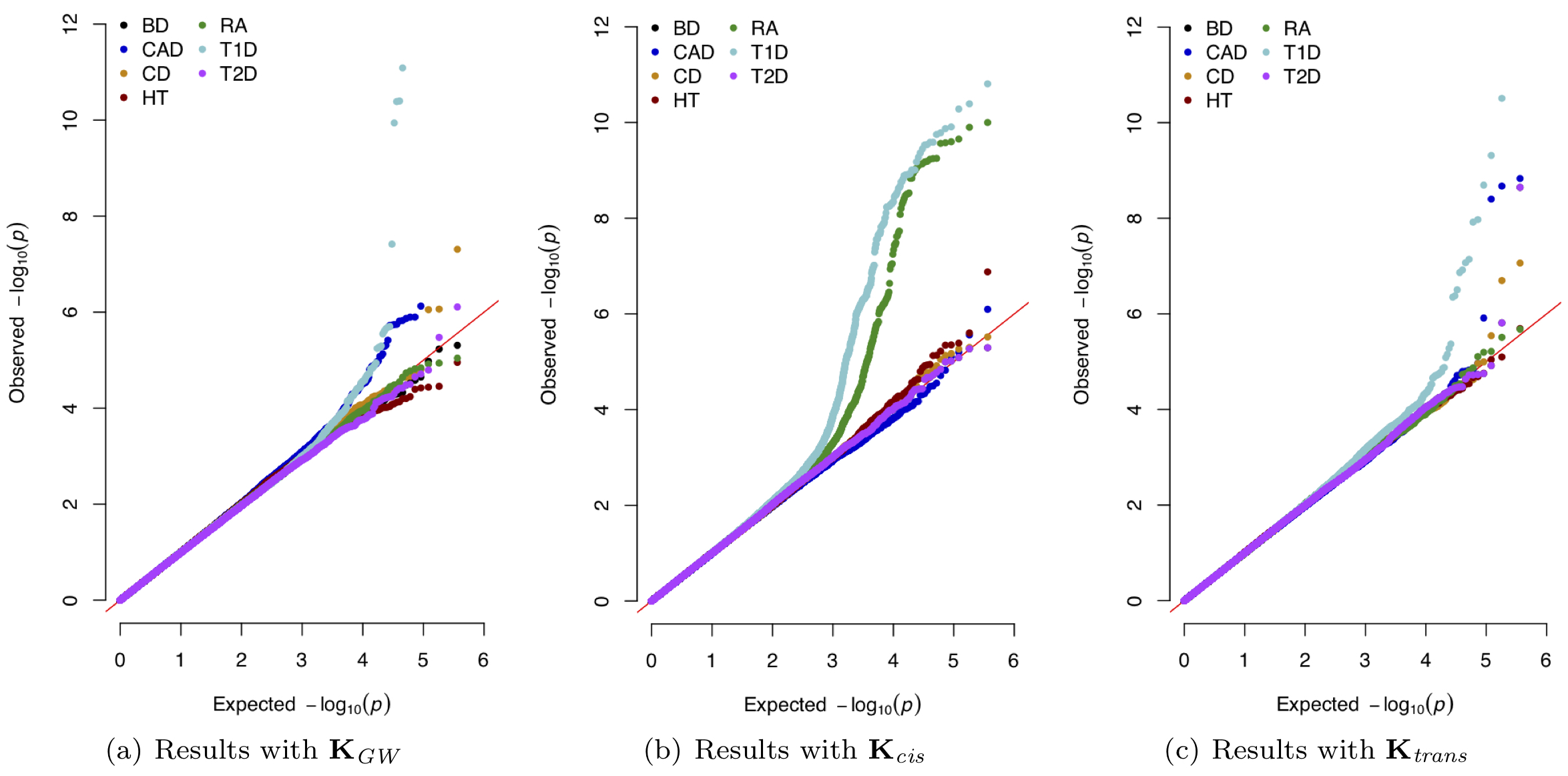
- M.C. Turchin#, G. Darnell, L. Crawford#, and S. Ramachandran#. Pathway analysis within multiple human ancestries reveals novel signals for epistasis in complex traits. bioRxiv. 2020.09.24.312421. [Preprint] [Software]
- W. Cheng, G. Darnell, S. Ramachandran, and L. Crawford#. Generalizing variational autoencoders with hierarchical empirical Bayes. arXiv. 2007.10389. [Preprint] [Software]
- K.E. Ware, S. Gupta, J. Eng, G. Kemeny, B.J. Puviindran, W.C. Foo, L. Crawford, R.G. Almquist, D. Runyambo, B.C. Thomas, M.U. Sheth, A. Agarwal, M. Pierobon, E.F. Petricoin, D.L. Corcoran, J. Freedman, S.R. Patierno, T. Zhang, S. Gregory, Z. Sychev, J.M. Drake, A.J. Armstrong#, and J.A. Somarelli#. Convergent evolution of p38/MAPK activation in hormone resistant prostate cancer mediates pro-survival, immune evasive, and metastatic phenotypes. bioRxiv. 2020.04.22.050385. [Preprint]
- J. Ish-Horowicz*, D. Udwin*, K. Scharfstein, S.R. Flaxman, L. Crawford#, and S.L. Filippi#. Interpreting deep neural networks through variable importance. arXiv. 1901.09839. [Preprint] [Software]
- L. Crawford# and X. Zhou#. Genome-wide marginal epistatic association mapping in case-control studies. bioRxiv. 374983. [Preprint] [SI] [Software]
2024:
- E.T. Winn-Nuñez#, M. Griffin, and L. Crawford# (2024). A simple approach for local and global variable importance in nonlinear regression models. Computational Statistics & Data Analysis. 194: 107914. [PDF] [Link] [SI] [Software]
2023:
- H. Adam#, F. Yin, M. Hu, N. Tenenholtz, L. Crawford, L. Mackey, and A. Koenecke (2023). Should I stop or should I go: early stopping with heterogeneous populations. Advances in Neural Processing Systems (NeurIPS). (Spotlight Paper) [Preprint] [Software]
- J. Stamp#, A. DenAdel, D. Weinreich, and L. Crawford# (2023). Leveraging the genetic correlation between traits improves the detection of epistasis in genome-wide association studies. G3: Genes, Genomes, Genetics. 13(8): jkad118. [PDF] [Software] [Documentation]
- C. Rios-Martinez, N. Bhattacharya, A.P. Amini, L. Crawford, and K.K. Yang# (2023). Deep self-supervised learning for biosynthetic gene cluster detection and product classification. PLOS Computational Biology. 19(5): e1011162. [PDF] [Software]
- A. Conard, A. DenAdel, and L. Crawford# (2023). A spectrum of explainable and interpretable machine learning approaches for genomic studies. WIREs Computational Statistics. 15(5): e1617. [PDF] [Link]
2022:
- B. Trippe#, B. Huang, E.A. DeBenedictis, B. Coventry, N. Bhattacharya, K.K. Yang, D. Baker, and L. Crawford# (2022). Randomized gates eliminate bias in sort-seq assays. Protein Science. 31(9): e4401. [PDF] [Link]
- W. Cheng#, S. Ramachandran, and L. Crawford# (2022). Uncertainty quantification in variable selection for genetic fine-mapping using Bayesian neural networks. iScience. 25(7): 104553. (Spotlight Talk at the 10th RECOMB Satellite on Computational Methods in Genetics) [PDF] [SI] [Software]
- W.S. Tang*, G.M. da Silva*, H. Kirveslahti, E. Skeens, B. Feng, T. Sudijono, K.K. Yang, S. Mukherjee, B. Rubenstein†, and L. Crawford†# (2022). A topological data analytic approach for discovering biophysical signatures in protein dynamics. PLOS Computational Biology. 18(5): e1010045. [PDF] [SI] [Software]
- S.P. Smith, S. Shahamatdar, W. Cheng, S. Zhang, J. Paik, M. Graff, C. Haiman, T.C. Matise, K.E. North, U. Peters, E. Kenny, C. Gignoux, G. Wojcik, L. Crawford†, and S. Ramachandran†# (2022). Enrichment analyses identify shared associations for 25 quantitative traits in over 600,000 individuals from seven diverse ancestries. American Journal of Human Genetics. 109: 871-884. [PDF] [Software]
2021:
- S. Raghavan*, P.S. Winter*#, A.W. Navia*, H.L. Williams*, A. DenAdel, R.L. Kalekar, J. Galvez-Reyes, K.E. Lowder, J. Galvez-Reyes, R.L. Kalekar, N. Mulugeta, K.S. Kapner, M.S. Raghavan, A.A. Borah, N. Liu, S.A. Väyrynen, A. Dias Costa, R.W.S. Ng, J. Wang, E.K. Hill, D.Y. Ragon, L.K. Brais, A.M. Jaeger, L.F. Spurr, Y.Y. Li, A.D. Cherniack, M.A. Booker, E.F. Cohen, M.Y. Tolstorukov, I. Wakiro, A. Rotem, B.E. Johnson, J.M. McFarland, E.T. Sicinska, T.E. Jacks, R.J. Sullivan, T.E. Clancy, K. Perez, D.A. Rubinson, K. Ng, J.M. Cleary, L. Crawford, S.R. Manalis, J.A. Nowak, B.R. Wolpin†, W.C. Hahn†, A.J. Aguirre†#, and A.K. Shalek†# (2021). Microenvironment drives cell state, plasticity, and drug response in pancreatic cancer. Cell. 184(25): 6119-6137. [PDF] [Link]
- M. Kamariza#, L. Crawford#, D. Jones#, and H.K. Finucane# (2021). Misuse of the term "trans-ethnic" in genomics research. Nature Genetics. 50: 1520-1521. [Link] [Editorial]
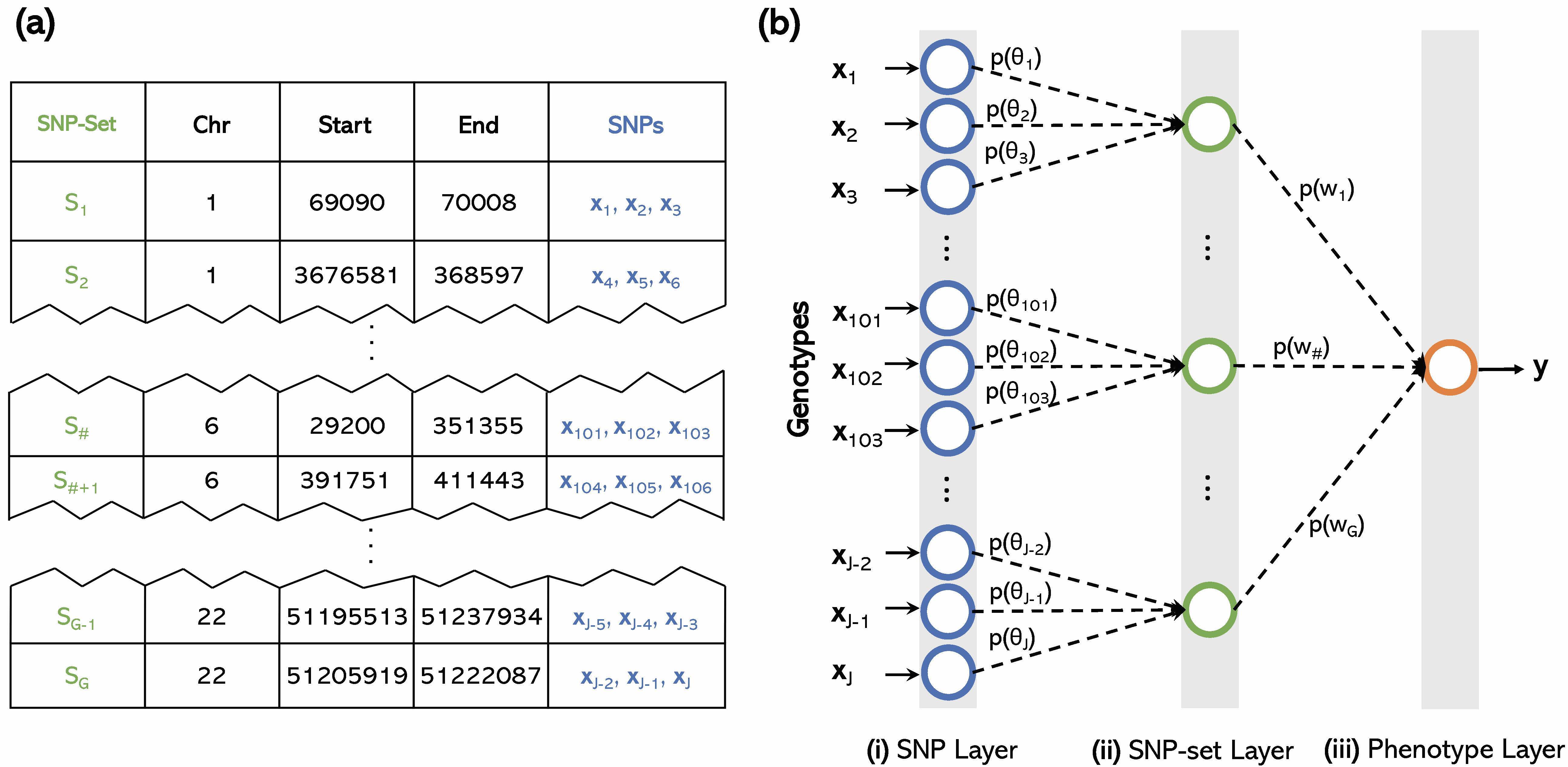
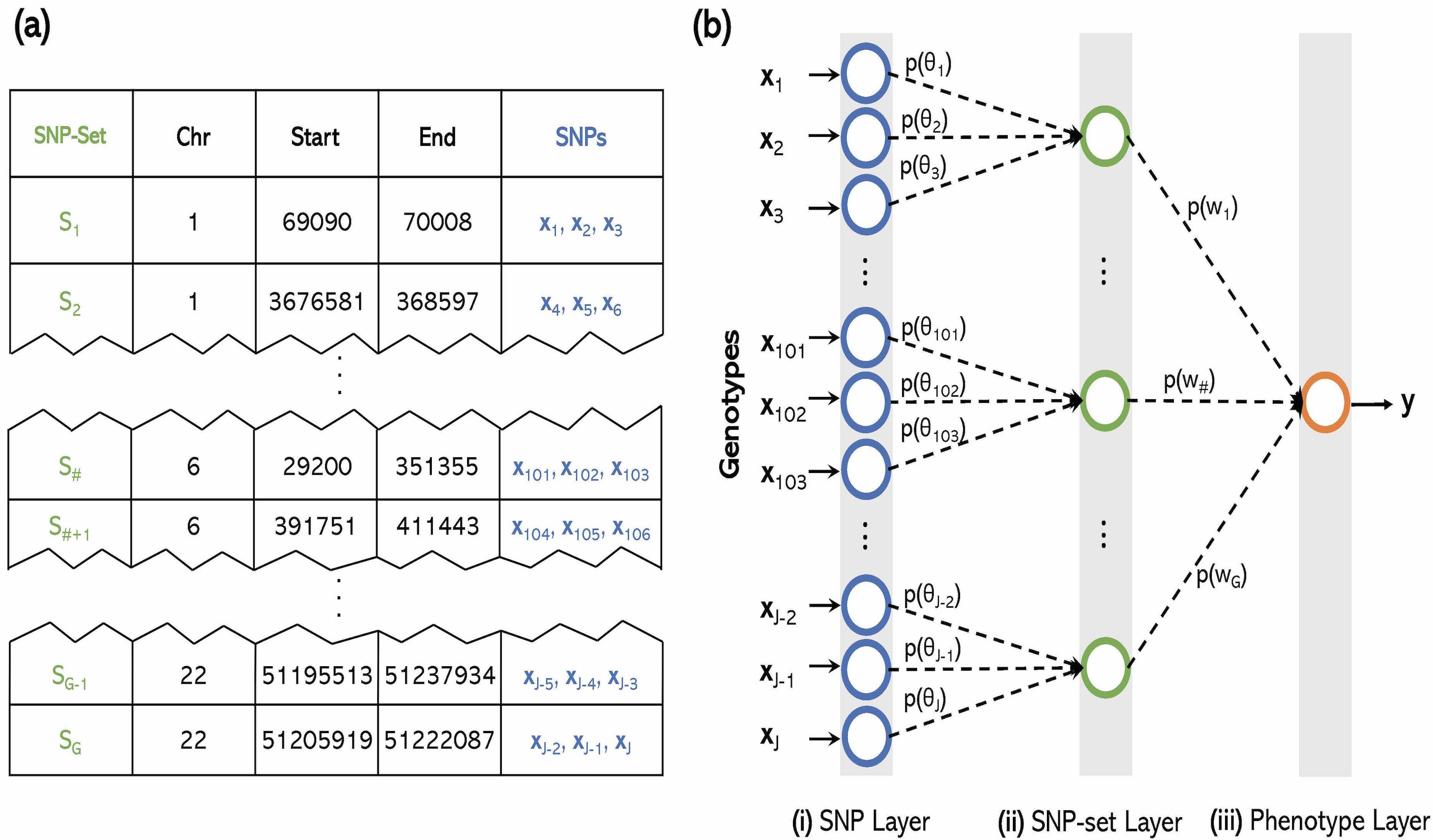
- P. Demetci*, W. Cheng*, G. Darnell, X. Zhou, S. Ramachandran, and L. Crawford# (2021). Multi-scale inference of genetic architecture using biologically annotated neural networks. PLOS Genetics. 17(8): e1009754. [PDF] [SI] [Software]
- D.E. Runcie#, J. Qu, H. Cheng, and L. Crawford (2021). Mega-scale linear mixed models for genomic predictions with thousands of traits. Genome Biology. 22: 213. [PDF] [SI] [Software]
- B. Wang*, T. Sudijono*, H. Kirveslahti*, T. Gao, D.M. Boyer, S. Mukherjee†, and L. Crawford†# (2021). A statistical pipeline for identifying physical features that differentiate classes of 3D shapes. Annals of Applied Statistics. 15(2): 638-661. [PDF] [SI] [Software]
- A.N. Spierer#, J.A. Mossman, S.P. Smith, L. Crawford, S. Ramachandran, and D.M. Rand# (2021). Natural variation in the regulation of neurodevelopmental genes modifies flight performance in Drosophila. PLOS Genetics. 17(3): e1008887. [PDF] [SI] [Software]
- B.A. Borden, Y. Baca, J. Xiu, F. Tavora, I. Winer, B.A. Weinberg, A.M. VanderWalde, S. Darabi, W.M. Korn, A.P. Mazar, F.J. Giles, L. Crawford, H. Safran, W.S. El-Deiry, and B.A. Carneiro# (2021). The landscape of glycogen synthase kinase-3 beta (GSK-3b) genomic alterations in cancer. Molecular Cancer Therapeutics. 20(1): 183-190. [Link]
2020:
- L. Crawford#, A. Monod#, A.X. Chen, S. Mukherjee, and R. Rabadán (2020). Predicting clinical outcomes in glioblastoma: an application of topological and functional data analysis. Journal of the American Statistical Association. 115(531): 1139-1150. [PDF] [SI] [Software]
- J.S. Sadick, L. Crawford, H.C. Cramer, C. Franck, S.A. Liddelow, and E.M. Darling# (2020). Generating cell type-specific protein signatures from non-symptomatic and diseased tissues. Annals of Biomedical Engineering. 48: 2218-2232. [Link]
- W. Cheng, S. Ramachandran#, and L. Crawford# (2020). Estimation of non-null SNP effect size distributions enables the detection of enriched genes underlying complex traits. PLOS Genetics. 16(6): e1008855. [PDF] [SI] [Software]
- K.H. Lin*, J.C. Rutter*, A. Xie, E.T. Winn, B. Pardieu, R. Dal Bello, Y.R. Ahn, Z. Dai, R.T. Sobhan, G.R. Anderson, K.R. Singleton, A.E. Decker, P.S. Winter, J.W. Locasale, L. Crawford, A. Puissant#, and K.C. Wood# (2020). Using antagonistic pleiotropy to design a chemotherapy-induced evolutionary trap. Nature Genetics. 52: 408-417. [PDF]
2019:
- T. Borgovan#, L. Crawford, C. Nwizu, and P. Quesenberry (2019). Stem cells and extracellular vesicles: biological regulators of physiology and disease. American Journal of Physiology-Cell Physiology. 317(2): C155-C166. [PDF]
- L. Crawford#, S.R. Flaxman, D.E. Runcie, and M. West (2019). Variable prioritization in nonlinear black box methods: a genetic association case study. Annals of Applied Statistics. 13(2): 958-989. [PDF] [SI] [Software]
- A. Monod#, S. Kališnik Verovšek, J.Á. Patiño-Galindo, and L. Crawford (2019). Tropical sufficient statistics for persistent homology. SIAM Journal on Applied Algebra and Geometry. 3(2): 337-371. [PDF] [Software]
- D.E. Runcie# and L. Crawford (2019). Fast and general-purpose linear mixed models for genome-wide genetics. PLOS Genetics. 15(2): e1007978. [PDF] [SI] [Software]
2018:
- L. Crawford#, K.C. Wood, X. Zhou#, and S. Mukherjee# (2018). Bayesian approximate kernel regression with variable selection. Journal of the American Statistical Association. 113(524): 1710-1721. [PDF] [SI] [Software]
- R. Soderquist, L. Crawford, E. Liu, M. Lu, A. Agarwal, G.R. Anderson, K.H. Lin, P.S. Winter, M. Cakir, and K.C. Wood# (2018). Systematic mapping of BCL-2 gene dependencies in cancer reveals molecular determinants of BH3 mimetic sensitivity. Nature Communications. 9(1): 3513. [PDF]
2017:
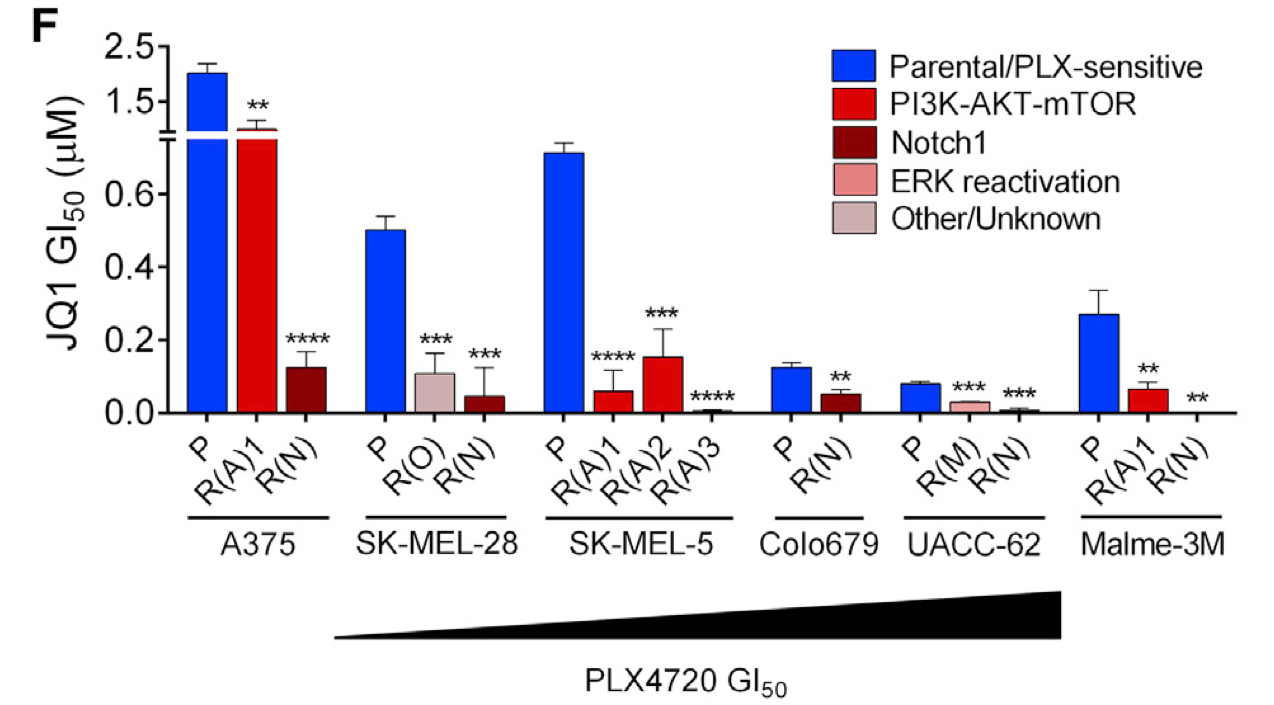
- K.R. Singleton*, L. Crawford*, E. Tsui, H.E. Manchester, O. Maertens, X. Liu, M.V. Liberti, A.N. Magpusao, E.M. Stein, J.P. Tingley, D.T. Frederick, G.M. Boland, K.T. Flaherty, S.J. McCall, C. Krepler, K. Sproesser, M. Herlyn, D.J. Adams, J.W. Locasale, K. Cichowski, S. Mukherjee, and K.C. Wood (2017). Melanoma therapeutic strategies that select against resistance by exploiting MYC-driven evolutionary convergence. Cell Reports. 21(10): 2796-2812. [PDF] [SI]
- L. Crawford#, P. Zeng, S. Mukherjee, and X. Zhou# (2017). Detecting epistasis with the marginal epistasis test in genetic mapping studies of quantitative traits. PLOS Genetics. 13(7): e1006869. [PDF] [SI] [Software]
- G.R. Anderson*, P.S. Winter*, K.H. Lin, D.P. Nussbaum, M. Cakir, E.M. Stein, R. Soderquist, L. Crawford, J.C. Leeds, R. Newcomb, P. Stepp, C. Yip, S.E. Wardell, J.P. Tingley, M. Ali, M. Xu, M. Ryan, S.J. McCall, A. McRee, C.M. Counter, C.J. Der, and K.C. Wood# (2017). A landscape of therapeutic cooperativity in KRAS mutant cancers reveals principles for controlling tumor evolution. Cell Reports. 20(4): 999-1015. [PDF]
2016:
- G.R. Anderson, S.E. Wardell, M. Cakir, L. Crawford, J.C. Leeds, D.P. Nussbaum, P.S. Shankar, R.S. Soderquist, E.M. Stein, J.P. Tingley, P.S. Winter, E.K. Zeiser-Misenheimer, H.M. Alley, A. Yllanes, V. Haney, K.L. Blackwell, S.J. McCall, D.P. McDonnell, and K.C. Wood# (2016). PIK3CA mutations enable selective targeting of a breast tumor lineage survival dependency through MTOR-mediated control of MCL-1 translation. Science Translational Medicine. 8: 369ra175. [PDF]
2014-2015:
- L. Crawford, V. Ponomarenko#, J. Steinberg, and M. Williams (2014). Accepted elasticity in local arithmetic congruence monoids. Results in Mathematics.66:227-245. [Link]
Stay Connected
-
Address
1 Memorial Dr.
Cambridge, MA 02142
United States
-
Phone
857-453-6156
-
Email
lcrawford (at) microsoft (dot) com
-
Email
lorin_crawford (at) brown (dot) edu